

Arbeitsgruppe von Hesperos in Orlando, Florida. Hintere Reihe in der Mitte: Chief Scientist Prof. Dr. James Hickman.
Foto: Hesperos Inc.
In Deutschland leiden rund vier Millionen Patienten an einer seltenen Krankheit. Nur die wenigsten von ihnen profitieren von einer medikamentösen Therapie. Ein Grund dafür ist, dass die Wirkmechanismen bei seltenen Krankheiten oft unbekannt sind. Solche Krankheiten werden traditionell an Mäusen, seltener an Zebrafischen oder Ratten erforscht. Doch die Tiere entwickeln solche Krankheiten gar nicht, sondern in einem Tier kann nur ein ähnliches Krankheitsphänomen gentechnisch erzeugt werden. Dementsprechend werden selten neue Behandlungsmethoden gefunden. Organ- und Human-on-a-Chip-Entwicklungen wie die des Start-ups Hesperos Inc. machen vor allem deshalb Sinn, weil die Entwicklung von Tiermodellen teuer ist und die Kosten für die Entwicklung eines Medikaments aufgrund der geringen Patientenzahl sehr hoch sind.
Seltene Erkrankungen
Es wird geschätzt, dass europaweit etwa 30 Millionen Patienten an einer seltenen Krankheit leiden. Eine Krankheit gilt als selten, wenn sie nicht mehr als fünf Menschen pro 10.000 Einwohner betrifft; in den USA darf die Gesamtzahl 200.000 nicht überschreiten. 6.000 bis 8.000 seltene Krankheiten sind bekannt, wobei sich die Wissenschaftler über die Zahl nicht einig sind (1). Die schlechte Nachricht: Nur für gut 100 der 8.000 Erkrankungen gibt es eine medikamentöse Therapie (2). Die schlechte Nachricht ist, dass es nur für 100 der 8.000 Krankheiten eine medikamentöse Therapie gibt (2). Einer der Gründe dafür ist, dass die Krankheitsmechanismen oft unbekannt sind. Seltene Krankheiten sind extrem vielfältig und komplex. Es kann sich um bestimmte Formen von Krebs, Erkrankungen des Herz-Kreislauf-Systems, der Nerven, des Stoffwechsels oder um Autoimmunerkrankungen handeln (3). Da sie überwiegend genetischen Ursprungs sind, können sie bereits im Kindesalter auftreten, führen zu gesundheitlichen Beeinträchtigungen und verkürzen häufig die Lebenserwartung (2). Beispiel: Die Neuronale Ceroid-Lipofuszinose (NCL) bündelt eine Gruppe von neurodegenerativen Erkrankungen mit ähnlichen klinischen Symptomen wie Demenz, Sehverlust sowie Epilepsie im Kindes- und Jugendalter. Typische Anzeichen der Erkrankung treten zwischen dem zweiten und vierten Lebensjahr auf (4). Dabei lagert sich in den Zellen des ZNS eine fettige, bräunliche Substanz (Ceroidlipofuszin) ab, die zum Absterben neuronaler Zellen führt (5, 6). Solche Zellen des Zentralnervensystems können zu einem patientenspezifischen Modell entwickelt werden, das alle Merkmale der Krankheit trägt, um Medikamentenwirkungen zu erforschen.
Patienten mit der seltenen Erkrankung Neuronale Ceroid-Lipofuszinose (NCL) sind oft schon im späten Kindesalter auf einen Rollstuhl angewiesen.
Fotoquelle: iStockphoto.
Weil die Erkrankungen der Patienten so selten sind, sind Pharmaunternehmen unter normalen Marktbedingungen nicht bereit, in diese Forschung zu investieren. Daher sind Anreize nötig, um erfolgreich Medikamente zu entwickeln (7). Ermutigt werden die Medikamentenentwickler durch die europäische Verordnung zu Orphan Drugs, die seit dem Jahr 2000 in Kraft ist. In den USA gibt es seit 1983 den Orphan Drug Act (ODA), der ebenfalls Anreize für die Erforschung vernachlässigter Krankheiten bietet, die sonst die Entwicklungskosten nicht decken würden (8). Deutschland hat im August 2013 einen Nationalen Aktionsplan für seltene Krankheiten verabschiedet (1). In der Europäischen Union (EU) sind derzeit 121 Medikamente für die Behandlung seltener Krankheiten verfügbar (9).
Umstrittene Tiermodelle zur Erforschung seltener Erkrankungen
Die Grundlagen seltener Erkrankungen sowie mögliche Arzneimittel werden traditionell mit Mäusen untersucht, seltener mit Zebrafischen oder Ratten. Ist eine Substanz gefunden, so folgen gesetzlich vorgeschriebene Tierversuche unter anderem auch mit nicht-humanen Primaten (z.B. Javaneraffen). Die Grundlagen seltener Krankheiten sowie mögliche Medikamente werden traditionell mit Mäusen, seltener mit Zebrafischen oder Ratten untersucht. Ist eine Substanz gefunden, folgen gesetzlich vorgeschriebene Tierversuche, auch mit nichtmenschlichen Primaten (z. B. Javaneraffen).
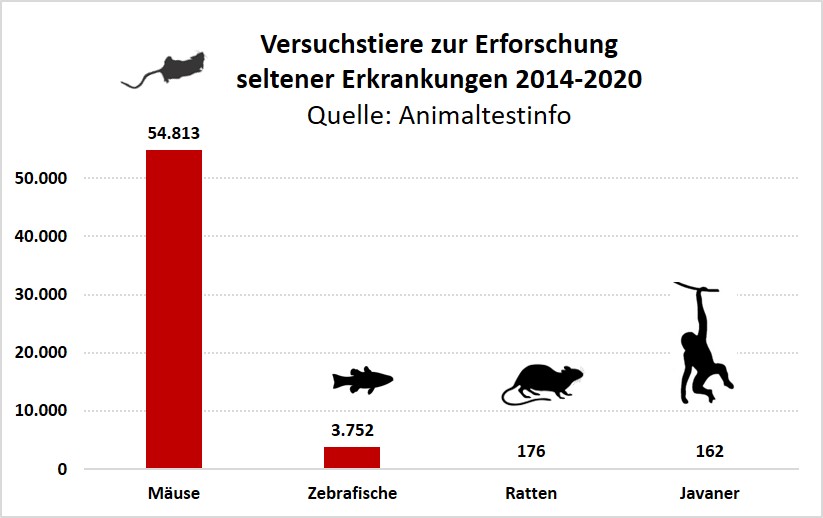
Hauptsächlich werden gentechnisch Mäuse für die Erforschung seltener Erkrankungen eingesetzt. Nicht-humane Primaten (Javaneraffen) werden in gesetzlich vorgeschriebenen Tierversuchen der präklinischen Entwicklung von Arzneimitteln eingesetzt.
Quelle: www.Animaltestinfo.de (10)
Allerdings können Tiermodelle die im menschlichen Körper beobachteten Reaktionen auf Medikamente aufgrund von Speziesunterschieden oft nicht nachbilden. Tiere entwickeln diese Krankheiten gar nicht; es kann nur ein ähnliches Krankheitsphänomen im Tier gentechnisch erzeugt werden - mit entsprechenden Ungenauigkeiten. Tierversuche sind zudem oft mit schwerem Stress für die Tiere verbunden. Die Sandhoff-Krankheit zum Beispiel beruht auf einer Mutation am HEXB-Gen, die dazu führt, dass zwei Untereinheiten eines wichtigen Enzyms (Hexosaminidase) fehlen oder mangelhaft sind. In der Folge reichern sich wasserunlösliche Fette vor allem in den Nervenzellen an. Die Folge ist eine Stagnation in der Entwicklung des Kindes, wobei erlernte Fähigkeiten später wieder verloren gehen. Es kann auch zur Erblindung kommen. Betroffene Kinder sterben meist im Alter von 4 Jahren (11, 12).
Zur Erforschung der hirnendothelspezifischen Gentherapie wird ein gentechnisch verändertes Mausmodell der Sandhoff-Krankheit verwendet. Allerdings ist es in der wissenschaftlichen Gemeinschaft nicht unumstritten: Die gentechnisch veränderte (hexb-/-) Sandhoff-Maus zeigt z. B. schwere motorische Funktionsstörungen und eine kurze Lebenserwartung, was im Gegensatz zum Menschen durch einen alternativen Abbauweg von GM2-Gangliosid im Mausmodell erklärt werden kann. Außerdem wird hier im Gegensatz zum Menschen eine Hyperaktivität des Immunsystems mitverantwortlich gemacht (13, 14, 15, 16).
Eine Chance für tierversuchsfreie Verfahren
Für Entwickler von Organ-on-a-Chip- oder Human-on-a-Chip-Modellen ist die Simulation seltener Krankheiten eine Chance. Denn sie hat das Potenzial, patientenindividuelle Behandlungsmöglichkeiten zu entdecken, was Kosten und Zeit reduzieren sowie die Zulassungsrate von Medikamenten erhöhen kann (17). In einzelnen Fällen ist dies bereits gelungen: Die seltene Kardiomyopathie Leigh-Syndrom konnte bereits erfolgreich behandelt werden, nachdem an einem personalisierten In-vitro-Krankheitsmodell ein geeignetes Medikament gefunden wurde (18, 19, 20).
Wissenschaftler von Hesperos untersuchten kürzlich die Amyotrophe Lateralsklerose (ALS) unter Verwendung von aus induzierten pluripotenten Stammzellen (iPSC) abgeleiteten Motoneuronen, um eine ganzheitliche Behandlung in einem neuromuskulären Kreuzungsmodell zu evaluieren (21). Die Amyotrophe Lateralsklerose (ALS) ist eine fortschreitende neurodegenerative Erkrankung, die durch einen selektiven Verlust motorischer Neuronen im Rückenmark und in den Hirnnervenkernen des Hirnstamms und des motorischen Kortex des Gehirns gekennzeichnet ist (24). Es kommt zu Muskelatrophie, Muskelschwäche, Spastiken und Lähmungen, die zum Tod durch fortschreitende Atrophie der Atemmuskulatur führen. Die Ursache der Krankheit ist, mit Ausnahme seltener erblicher Formen, noch unbekannt.
In einer weiteren Studie verwendeten die Hesperos-Wissenschaftler ein In-vitro-Modell mit menschlichen Skelettmuskeln auf einem Muskelfunktions-Chip. Die Skelettmuskeln wurden aus induzierten pluripotenten Stammzellen von ALS-Patienten entwickelt. Die Wissenschaftler verglichen die Eigenschaften der Skelettmuskeln der Patienten mit denen von gesunden Probanden. Die Skelettmuskeln der ALS-Patienten hatten Mutationen in einem Gen, das für ein wichtiges Enzym (Superoxid-Dismutase 1) kodiert. In ihrem Modell konnten sie zeigen, dass dies zu einer verminderten Fusionsfähigkeit des Muskels, einer Verkürzung der Myotubuslänge und der verminderten Bildung wichtiger Acetylcholinrezeptoren führt. ALS-Muskeln zeigten beispielsweise auch eine verminderte kontraktile Kraft und einen gestörten Muskelfaserstoffwechsel (21).
Hesperos Inc. hat die Erforschung seltener Krankheiten zu einem seiner Hauptschwerpunkte gemacht. Das in Orlando ansässige Start-up-Unternehmen entwickelt pumpenfreie mikrophysiologische Multi-Organ-Systeme (Human-on-a-Chip-Systeme), die für die Erforschung seltener Krankheiten geeignet sind, da sie für einzelne Krankheiten maßgeschneidert werden können. Gegründet wurde das Unternehmen von den Professoren Dr. Michael Shuler und Dr. James J. Hickman, die über langjährige Erfahrung in der Body-on-a-Chip-Forschung verfügen, wobei Dr. Shuler als Erfinder dieses Bereichs gilt. Hesperos hat bereits mikrofluidische Systeme mit menschlichen Herz-, Muskel-, Glia-, Endothel-, Hepatozyten-, Knochenmark-, Krebs-, Immun- und Epithelzellen gebaut und Validierungsstudien mit der Industrie durchgeführt (22). Das Design ihrer mikrofluidischen Chipsysteme ist pumpenlos, um den Bedarf an Pumpen und Schläuchen zu eliminieren, was die Plattformen vereinfacht, die Blasenbildung eliminiert und es zu einem System mit geringem Volumen macht, das die Analyse sowohl der Mutterzelle als auch ihrer Metaboliten ermöglicht, wenn ein Lebermodell einbezogen wird.
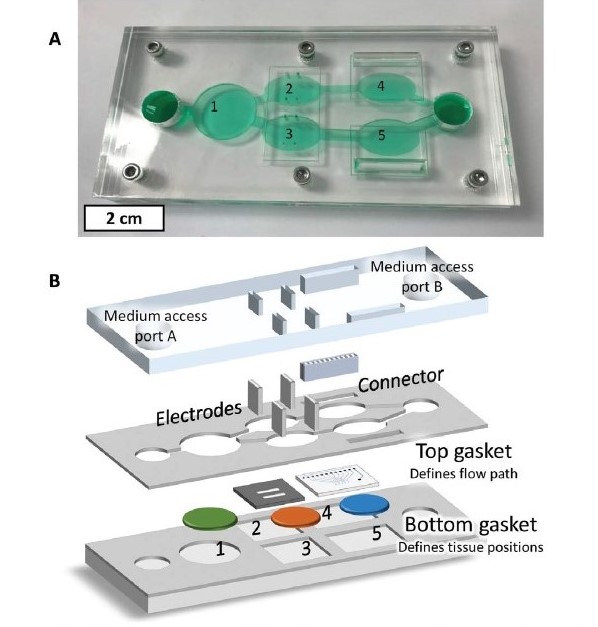
Beispiel für ein entwickeltes Multiorgan-System.
A) 5-Kammer-Multiorgan-System in der Übersicht.
B) Kammer 1 enthält Leberzellen, Kammer 2 und 4 sind Herzzellen auf Cantilevern bzw. auf Multielektrodenarrays (MEAs), Kammern 3 und 5 wurden in diesem Fall für zwei unterschiedliche Krebszelllinien verwendet. Testsubstanzen werden dem Medium in Port A hinzugegeben und durchlaufen als erstes die Leber, um den first-Pass-Metabolismus zu simulieren.
Quelle: McAleer et al. (2019). Science Translational Medicine 11/497, DOI: 10.1126/scitranslmed.aav1386.
(Mit freundlicher Genehmigung von AAAS.)**
Ein Beispiel für die Fülle der Entwicklungen des Start-ups Hesperos ist der sogenannte Cantilever-Chip: Er besteht aus 32 mikroskopisch kleinen Silizium-Auslegern, auf deren Oberfläche Herz- oder Muskelzellen kultiviert werden können. Mit einem Laser- und Photodetektorsystem wird die Zellkontraktion an den Cantilevern aufgezeichnet, die bei der kleinsten Zellbewegung in Beugung versetzt werden. Wenn eine Testsubstanz auf eine Muskelzelle einwirkt, wird die Kontraktionsabweichung in Größe, Zeit und Dauer genau erfasst. In einem kombinierten Modell werden Multielektroden-Arrays zur Messung der elektrischen Aktivität von Zellen, vor allem von Herz- und Nervenzellen, und das Cantilever-Modell zur Messung von Stresskontraktionen von Herzmuskelzellen sowie von Skelettmuskeln mit dem Photodiodenlaser- und Detektorsystem verwendet (23).
"... eine einfache, effektive und auch relativ kostengünstige Möglichkeit, seltene Krankheiten zu erforschen..."
Die Erforschung seltener Krankheiten ist für Pharmaunternehmen unattraktiv. Wegen des hohen Aufwands und der geringen Zahl der betroffenen Patienten stehen Kosten und Nutzen für die Unternehmen einfach nicht im Verhältnis. Hinzu kommt, dass für die Forschung nur wenig Geld zur Verfügung steht - und wenig oder keine so genannten Tiermodelle, an denen man sie untersuchen kann. Deshalb ist das Forschungsgebiet der seltenen Krankheiten ein ideales Beispiel, um die Vorteile aufzuzeigen, die komplexe tierfreie Methoden wie die Human-on-a-Chip-Technologie bieten. Wir sprachen mit Chief Scientist Prof. Dr. James J. Hickman über das Potenzial dieser tierfreien Techniken für die Erforschung seltener Krankheiten und darüber hinaus.
InVitro+Jobs: Zur Erforschung von Krankheiten und Therapien werden immer noch hauptsächlich Tiermodelle eingesetzt. Wie ist die Situation bei seltenen Krankheiten?
Prof. Hickman: Es gibt über 7.700 bekannte seltene Krankheiten, denen nur etwa 400 aktive Forschungsprogramme gegenüberstehen. Das liegt daran, dass aufgrund der geringen Patientenzahl in den meisten Fällen nur minimale Mittel für die Erforschung seltener Krankheiten zur Verfügung stehen. Daher gibt es nur wenige Tiermodelle für seltene Krankheiten, und in vielen Fällen sind sie, weil es sich um humanisierte Modelle handelt, unerschwinglich teuer. Darüber hinaus gibt es für die meisten seltenen Krankheiten aus technischen Gründen überhaupt keine Tiermodelle.
InVitro+Jobs: Können Sie in einfachen Worten erklären, wie die Human-on-a-Chip-Technologie auf diesem Gebiet funktioniert?
Prof. Hickman: Es ist ein einfacher Prozess, Stammzellen von Patienten mit einer seltenen Krankheit zu erzeugen, die Zellen in die am stärksten betroffenen Zellen zu differenzieren oder Serumproben von Patienten zu verwenden, um phänotypische Modelle zu entwickeln, die sich vom gesunden Probanden (Wildtyp) in einigen Schlüsselfunktionen unterscheiden. Alle Moleküle*, die die Leistung verbessern und sie näher an den Wildtyp heranbringen können, hätten das Potenzial, die Krankheit zu behandeln.
InVitro+Jobs: Also ist die Wissenschaft in diesem Bereich praktisch gezwungen, diese Krankheiten mit tierfreien Methoden zu untersuchen?
Prof. Hickman: Die Human-on-a-Chip-Technologie ist eine einfache, effektive und relativ kostengünstige Möglichkeit, seltene Krankheiten zu erforschen. Das führt automatisch zu einer Reduzierung der Tierversuche in diesem Bereich durch die Chip-Technologie.
InVitro+Jobs: Könnte man im Umkehrschluss sagen, dass die Existenz von tierischen Krankheitsmodellen die Forschung und die effektive Entwicklung bestimmter Therapien sogar behindert? Denn wenn ein Tiermodell existiert, wird die Chiptechnologie gar nicht genutzt und kann sich als Methode nicht bewähren.
Prof. Hickman: Nein, selbst wenn ein Tiermodell existiert, kann die HoaC-Technologie helfen, diese Experimente zu reduzieren und zu verfeinern und sich während solcher Experimente möglicherweise als besseres Modell als die Versuchstiere erweisen. Wenn also ein Tiermodell existiert, schließt das die Anwendung einer HoaC-Technologie auf die seltene Krankheit nicht aus.
InVitro+Jobs: Welche seltenen Krankheiten können bereits mit der Human-on-a-Chip (HoaC) Technologie erforscht werden?
Prof. Hickman: Bei Hesperos haben wir eine Reihe von vielversprechenden Medikamentenkandidaten für verschiedene seltene Krankheiten, die mit der HoaC-Technologie erforscht wurden, und wir glauben, dass die Mehrzahl der seltenen Krankheiten mit der HoaC-Technologie angegangen werden kann. Wir haben bereits einen Antrag auf Zulassung eines neuen Medikaments für eine seltene Krankheit mit dieser Methode eingereicht und zahlreiche weitere in der Entwicklung.
InVitro+Jobs: Welche weiteren Vorteile bieten die HoaC-Methoden für die Erforschung seltener Krankheiten?
Prof. Hickman: Die meisten Tiermodelle für seltene Krankheiten existieren derzeit nicht oder sind aus technischen oder finanziellen Gründen nicht machbar. Außerdem ist es wahrscheinlich ein besseres Modell für die Erforschung der Wirksamkeit von Medikamenten im Vergleich zu anderen Techniken, weil wir phänotypische Modelle mit echten Patientenproben erstellen können. Das Aufkommen der Stammzelltechnologie macht es einfach, Patientenzellen in Stammzellen umzuwandeln, die dann zu den betroffenen Zellen differenziert werden können. Bei anderen seltenen Krankheiten wird nur Plasma aus Blutproben ausreichen, zum Beispiel bei einigen Immunkrankheiten, bei denen sich die Antikörper, die die Krankheit verursachen, im Blut befinden.
InVitro+Jobs: Welche seltenen Krankheiten können bei Hesperos bereits mit HoaC untersucht werden?
Prof. Hickman: Hesperos konzentriert sich derzeit hauptsächlich auf neurologische seltene Krankheiten, sowohl auf Erkrankungen des zentralen Nervensystems als auch des peripheren Nervensystems. Wir erforschen auch lysosomale Speicherkrankheiten sowie Leber- und Herzphänomene.
InVitro+Jobs: Könnte man sagen, dass mehr Mittel für die Chiptechnologie auch zu mehr Therapien für betroffene Patienten führen würden?
Prof. Hickman: Ja, ich glaube, das ist der Fall, vor allem, wenn die Förderung auf seltene Krankheiten ausgerichtet ist.
InVitro+Jobs: Wie werden Entwickler bei der Erforschung seltener Krankheiten unterstützt, z.B. durch den Orphan Drug Act (ODA)?
Prof. Hickman: Hesperos konnte sich zur Zeit nicht direkt bei diesem Programm bewerben. Es arbeitet aber mit Firmen zusammen, die diesen Weg genutzt haben, um Kapital zu beschaffen oder Mittel zu erhalten, die ihnen sonst für die Forschung an seltenen Krankheiten nicht zur Verfügung gestanden hätten.
InVitro+Jobs: Was sind die Schwierigkeiten bei der Entwicklung einer Chip-Technologie für seltene Krankheiten?
Prof. Hickman: Die Hauptbeschränkung bei der Erforschung seltener Krankheiten ist zur Zeit die Finanzierung und der Zugang zu Patientenproben. Es gibt auch einige seltene Krankheiten, bei denen der Einsatz der HoaC-Technologie nicht optimal ist. Aber da wir vernetzte Multi-Organ-Systeme individuell aufbauen können, haben wir ein breites Spektrum an Krankheitsindikationen, die wir angehen können.
InVitro+Jobs: Haben die Ergebnisse der Erforschung einer seltenen Krankheit mit HoaC bereits zur Entwicklung eines Medikaments geführt?
Prof. Hickman: Ich kann nicht für andere Firmen oder Forschungsanstrengungen auf dem Gebiet der seltenen Krankheiten sprechen, aber mir sind bisher keine Anstrengungen bekannt, die zu einem zugelassenen Medikament geführt haben. Das liegt aber nicht an der Leistungsfähigkeit der Technologie, sondern daran, dass sie erst seit kurzem kommerzialisiert wird. Die Erforschung seltener Krankheiten mit Hilfe von Chiptechnologien wächst rasant. Bei Hesperos haben wir eine Reihe von vielversprechenden Kandidaten für verschiedene seltene Krankheiten, und wir haben ein Molekül, an dem wir mit dem Pharmaunternehmen Bioverativ gearbeitet haben, das in einem unserer Funktionsmodelle hervorragende Ergebnisse erzielte. Das ermöglichte die Einreichung eines Zulassungsantrags für ein neues Medikament bei der FDA, der hauptsächlich auf diesen Ergebnissen basiert.
InVitro+Jobs: Haben die Entwicklungen zu Kooperationen mit der Pharmaindustrie geführt?
Prof. Hickman: Ja, wir haben Kooperationen mit kleinen, mittleren und großen Pharmaunternehmen sowie mit staatlichen, akademischen und gemeinnützigen Organisationen aufgebaut.
InVitro+Jobs: Gibt es an Ihrer Universität UCF bereits Kurse, in denen Studierende die In-vitro-Modellierung von seltenen Krankheiten erlernen können?
Prof. Hickman: Leider ist die Technologie noch zu neu, um bei der Verwaltung der UCF ein Interesse zu wecken, das die Einrichtung von Kursen auf diesem Forschungsgebiet ermöglichen würde.
InVitro+Jobs: Worin sehen Sie das Potenzial der Chip-Technologie über die Forschung an seltenen Krankheiten hinaus?
Prof. Hickman: Wir konzentrieren uns auf seltene Krankheiten, weil wir glauben, dass hier ein ungedeckter Bedarf besteht, aber wir und andere Firmen, die mikrophysiologische Systeme anbieten, arbeiten auch an allgemeineren Krankheiten wie Herzkrankheiten, Alzheimer und Diabetes.
InVitro+Jobs: Wir danken Ihnen für das Gespräch.